Welcome to MetaCerberus ReadTheDocs!
![]()
![]()






Note
Metacerberus version 1.3 is the newest version via manual install due to current Conda/Mamba issue.
About
MetaCerberus transforms raw sequencing (i.e. genomic, transcriptomics, metagenomics, metatranscriptomic) data into knowledge. It is a start to finish python code for versatile analysis of the Functional Ontology Assignments for Metagenomes (FOAM), KEGG, CAZy/dbCAN, VOG, pVOG, PHROG, COG, and a variety of other databases including user customized databases via Hidden Markov Models (HMM) for functional annotation for complete metabolic analysis across the tree of life (i.e., bacteria, archaea, phage, viruses, eukaryotes, and whole ecosystems). Metacerberus also provides automatic differential statistics using DESeq2/EdgeR, pathway enrichments with GAGE, and pathway visualization with Pathview R.

General Terminal Info and Help Links for Novices
The following are links to helpful webpages based on your operating system. These contain basic starter info for those who have no previous experience with terminals or commands.
Operating System
Linux
Here, you can find a tutorial covering the basics of the Linux command line, using Ubuntu.
Other informative pages can be found here and here.
Mac
Click here for terminal basics.
Windows - MUST use Ubuntu
Click here for the Ubuntu download page, or download in the Microsoft store.
Here, you can find a tutorial covering the basics of the Linux command line, using Ubuntu.
Installation
Installing MetaCerberus 1.3 manually due to Mamba/Conda issue (Newest Version)
Important
You still need to have Mamba and Conda installed. You cannot just use Mamba/Conda directly for the new version, currently. Click here for Conda download instructions. For each command given, enter the first line of the command, then press ENTER. Once the operation completes, the terminal prompt will reappear (blinking vertical line where you type). Proceed to the next line of the given command, press ENTER. Continue as such, line by line, until the entire given command has been entered.
In the command line, type:
git clone https://github.com/raw-lab/MetaCerberus.git
cd metacerberus
bash install_metacerberus.sh
conda activate MetaCerberus-1.3.0
metacerberus.py --download
Installing MetaCerberus 1.2.1 and below (due to current Mamba and Conda errors)
Note
We will update this as soon as Mamba/Conda corrects this error.
Option 1) Mamba
Note
Make sure to install Mamba in your base Conda environment unless you have OSX with ARM architecture (M1/M2 Macs). Follow the OSX-ARM instructions below if you have a Mac with ARM architecture.
Mamba install from bioconda with all dependencies:
Linux/OSX-64
Install Mamba using Conda
In command line, type:
conda install mamba
Install MetaCerberus with Mamba
In command line, type:
mamba create -n metacerberus -c bioconda -c conda-forge metacerberus
conda activate metacerberus
metacerberus.py --setup
OSX-ARM (M1/M2) [if using a Mac with ARM architecture]
Set up Conda environment
In command line, type:
conda create -y -n metacerberus
conda activate metacerberus
conda config --env --set subdir osx-64
Install Mamba, Python, and Pydantic inside the environment
In command line, type:
conda install -y -c conda-forge mamba python=3.10 "pydantic<2"
Install MetaCerberus with Mamba
In command line, type:
mamba install -y -c bioconda -c conda-forge metacerberus
metacerberus.py --setup
Note
Mamba is the fastest installer. Anaconda or miniconda can be slow. Also, install Mamba from Conda, NOT from pip. The Mamba from pip doesn’t work for install.
Option 2) Anaconda - Linux/OSX-64 Only
Anaconda install from bioconda with all dependencies:
In command line, type:
conda create -n metacerberus -c conda-forge -c bioconda metacerberus -y
conda activate metacerberus
metacerberus.py --setup
Overview
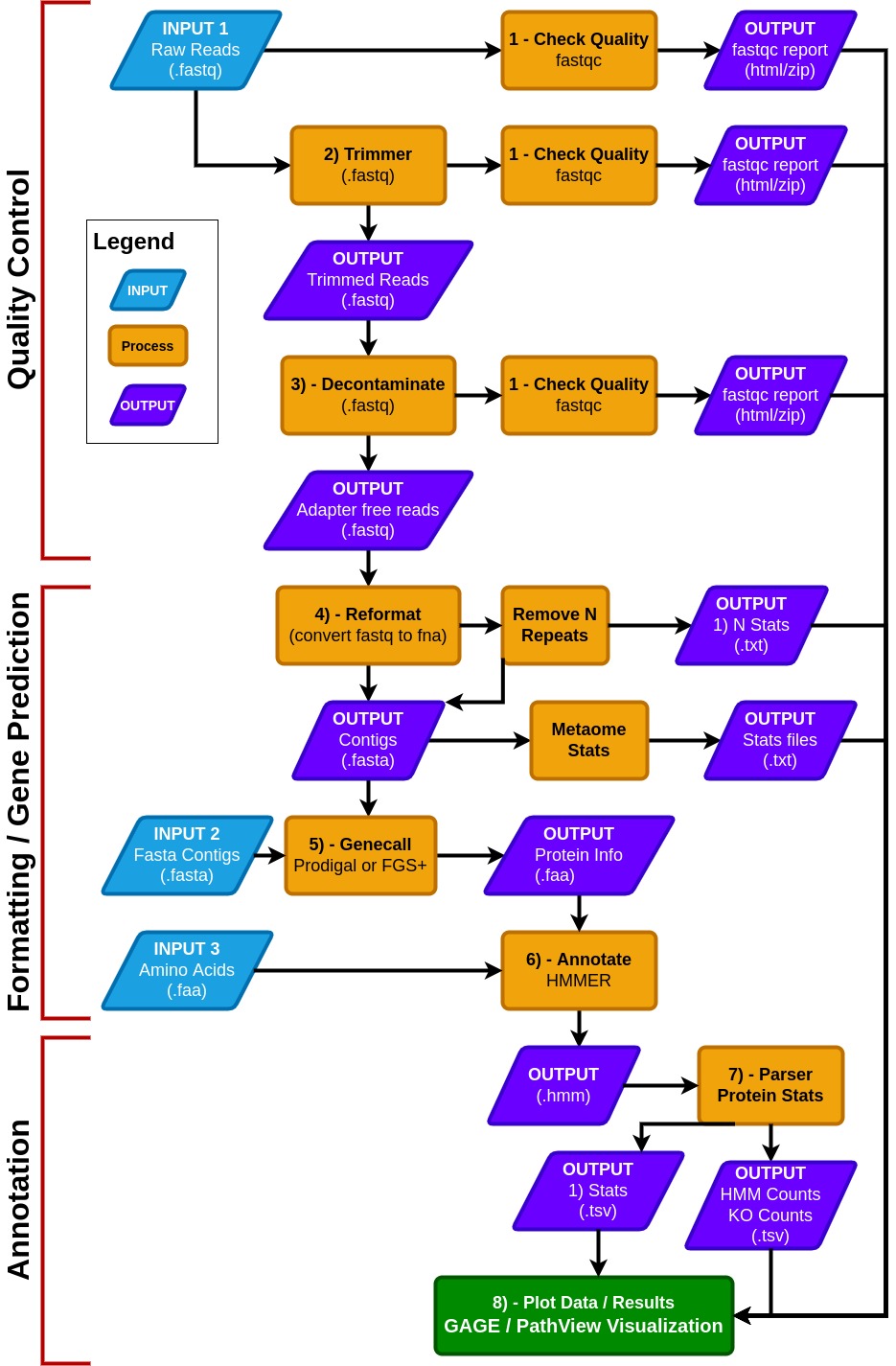
General Info
- MetaCerberus has three basic modes:
Quality Control (QC) for raw reads
Formatting/gene prediction
Annotation
- MetaCerberus can use three different input files:
Raw read data from any sequencing platform (Illumina, PacBio, or Oxford Nanopore)
Assembled contigs, as MAGs, vMAGs, isolate genomes, or a collection of contigs
Amino acid fasta (.faa), previously called pORFs
We offer customization, including running all databases together, individually or specifying select databases. For example, if a user wants to run prokaryotic or eukaryotic-specific KOfams, or an individual database alone such as dbCAN, both are easily customized within Metacerberus.
In QC mode, raw reads are quality controlled with pre- and post-trim via FastQC. Raw reads are then trimmed via data type; if the data is Illumina or PacBio, fastp is called, otherwise it assumes the data is Oxford Nanopore then PoreChop is utilized.
If Illumina reads are utilized, an optional bbmap step to remove the phiX174 genome is available or user provided contaminate genome. Phage phiX174 is a common contaminant within the Illumina platform as their library spike-in control. We highly recommend this removal if viral analysis is conducted, as it would provide false positives to ssDNA microviruses within a sample.
We include a
--skip_deconoption to skip the filtration of phiX174, which may remove common k-mers that are shared in ssDNA phages.In the formatting and gene prediction stage, contigs and genomes are checked for N repeats. These N repeats are removed by default.
We impute contig/genome statistics (e.g., N50, N90, max contig) via our custom module Metaome Stats.
Contigs can be converted to pORFs using Prodigal , FragGeneScanRs, and Prodigal-gv as specified by user preference.
Scaffold annotation is not recommended due to N’s providing ambiguous annotation.
Both Prodigal and FragGeneScanRs can be used via our
--superoption, and we recommend using FragGeneScanRs for samples rich in eukaryotes.FragGeneScanRs found more ORFs and KOs than Prodigal for a stimulated eukaryote rich metagenome. HMMER searches against the above databases via user specified bitscore and e-values or our minimum defaults (i.e., bitscore = 25, e-value = 1 x 10-9).
Input File Formats
From any NextGen sequencing technology (from Illumina, PacBio, Oxford Nanopore)
Type 1 raw reads (.fastq format)
Type 2 nucleotide fasta (.fasta, .fa, .fna, .ffn format), assembled raw reads into contigs
Type 3 protein fasta (.faa format), assembled contigs which genes are converted to amino acid sequence
Output Files
If an output directory is given, that folder will be created where all files are stored.
If no output directory is specified, the ‘results_metacerberus’ subfolder will be created in the current directory.
Gage/Pathview R analysis provided as separate scripts within R.
Visualization of Outputs
We use Plotly to visualize the data
Once the program is finished running, the html reports with the visuals will be saved to the _last_ step of the pipeline.
The HTML files require plotly.js to be present. One has been provided in the package and is saved to the report folder.
Annotation
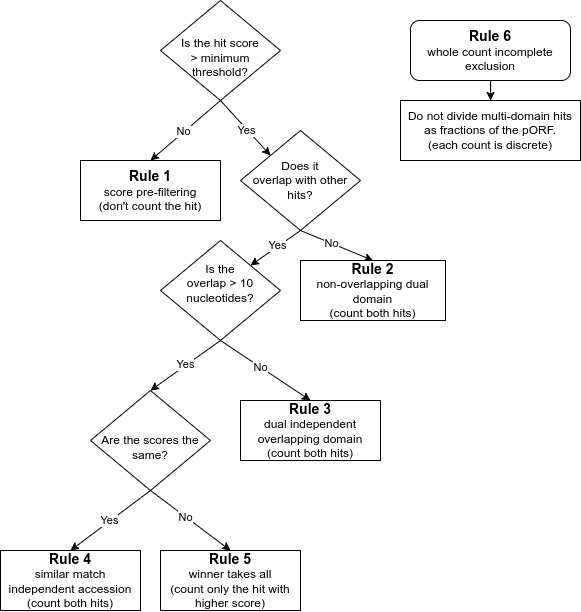
Rule 1 is for finding high quality matches across databases. It is a score pre-filtering module for pORFs thresholds: which states that each pORF match to an HMM is recorded by default or a user-selected cut-off (i.e., e-value/bit scores) per database independently, or across all default databases (e.g, finding best hit), or per user specification of the selected database.
Rule 2 is to avoid missing genes encoding proteins with dual domains that are not overlapping. It is imputed for non-overlapping dual domain module pORF threshold: if two HMM hits are non-overlapping from the same database, both are counted as long as they are within the default or user selected score (i.e., e-value/bit scores).
Rule 3 is to ensure overlapping dual domains are not missed. This is the dual independent overlapping domain module for convergent binary domain pORFs. If two domains within a pORF are overlapping <10 amino acids (e.g, COG1 and COG4) then both domains are counted and reported due to the dual domain issue within a single pORF. If a function hits multiple pathways within an accession, both are counted, in pathway roll-up, as many proteins function in multiple pathways.
Rule 4 is the equal match counter to avoid missing high quality matches within the same protein. This is an independent accession module for a single pORF: if both hits within the same database have equal values for both e-value and bit score but are different accessions from the same database (e.g., KO1 and KO3) then both are reported.
Rule 5 is the ‘winner take all’ match rule for providing the best match. It is computed as the winner takes all module for overlapping pORFs: if two HMM hits are overlapping (>10 amino acids) from the same database the lowest resulting e-value and highest bit score wins.
Rule 6 is to avoid partial or fractional hits being counted. This ensures that only whole discrete integer counting (e.g., 0, 1, 2 to n) are computed and that partial or fractional counting is excluded.
Quick Start Examples
Genome examples
All databases
conda activate metacerberus
metacerberus.py --prodigal lambda.fna --hmm ALL --dir_out lambda_dir
Only KEGG/FOAM all
conda activate metacerberus
metacerberus.py --prodigal lambda.fna --hmm KOFam_all --dir_out lambda_ko-only_dir
Only KEGG/FOAM prokaryotic centric
conda activate metacerberus
metacerberus.py --prodigal ecoli.fna --hmm KOFam_prokaryote --dir_out ecoli_ko-only_dir
Only KEGG/FOAM eukaryotic centric
conda activate metacerberus
metacerberus.py --fraggenescan human.fna --hmm KOFam_eukaryote --dir_out human_ko-only_dir
Only Viral/Phage databases
conda activate metacerberus
metacerberus.py --prodigal lambda.fna --hmm VOG, PHROG --dir_out lambda_vir-only_dir
Tip
You can pick any single database you want for your analysis including KOFam_all, COG, VOG, PHROG, CAZy or specific KO databases for eukaryotes and prokaryotes (KOFam_eukaryote or KOFam_prokaryote).
Custom HMM
conda activate metacerberus
metacerberus.py --prodigal lambda.fna --hmm Custom.hmm --dir_out lambda_vir-only_dir
Illumina data
Bacterial, Archaea and Bacteriophage metagenomes/metatranscriptomes
conda activate metacerberus
metacerberus.py --prodigal [input_folder] --illumina --meta --dir_out [out_folder]
Eukaryotes and Viruses metagenomes/metatranscriptomes
conda activate metacerberus
metacerberus.py --fraggenescan [input_folder] --illumina --meta --dir_out [out_folder]
Nanopore data
Bacterial, Archaea and Bacteriophage metagenomes/metatranscriptomes
conda activate metacerberus
metacerberus.py --prodigal [input_folder] --nanopore --meta --dir_out [out_folder]
Eukaryotes and Viruses metagenomes/metatranscriptomes
conda activate metacerberus
metacerberus.py --fraggenescan [input_folder] --nanopore --meta --dir_out [out_folder]
PacBio data
Microbial, Archaea and Bacteriophage metagenomes/metatranscriptomes
conda activate metacerberus
metacerberus.py --prodigal [input_folder] --pacbio --meta --dir_out [out_folder]
Eukaryotes and Viruses metagenomes/metatranscriptomes
conda activate metacerberus
metacerberus.py --fraggenescan [input_folder] --pacbio --meta --dir_out [out_folder]
SUPER (both methods)
conda activate metacerberus
metacerberus.py --super [input_folder] --pacbio/--nanopore/--illumina --meta --dir_out [out_folder]
Important
Fraggenescan will work for prokaryotes and viruses/bacteriophage but prodigal will not work well for eukaryotes.
Prerequisites and Dependencies
python >= 3.8
Available from Bioconda - external tool list
Tool |
Version |
Publication |
0.12.1 |
None |
|
0.23.4 |
||
0.2.4 |
None |
|
39.06 |
None |
|
2.6.3 |
||
v1.1.0 |
||
2.2.1 |
||
1.5.0 |
||
3.4 |
Metacerberus Databases
All pre-formatted databases are present at OSF.
Database sources
Note
The KEGG database contains KOs related to Human disease. It is possible that these will show up in the results, even when analyzing microbes. eggNOG and FunGene database are coming soon. If you want a custom HMM build please let us know by email or leaving an issue.
Custom Database
To run a custom database, you need a HMM containing the protein family of interest and a metadata sheet describing the HMM required for look-up tables and downstream analysis. For the metadata information you need an ID that matches the HMM and a function or hierarchy. See example below:
Example Metadata sheet
ID |
Function |
HMM1 |
Sugarase |
HMM2 |
Coffease |
Metacerberus Options
Important
If the Metacerberus environment is not used, make sure the dependencies are in PATH or specified in the config file.
Run
metacerberus.pywith the options required for your project.
Usage of metacerberus.py:
Note
The following are different options/arguments to modify the execution of Metacerberus.
Argument/Option |
Function [Default] |
Usage Format |
Accepted format |
Example (Type as one line) |
|
Setup additional dependencies [False] |
|
N/A |
|
|
Update downloaded databases [False] |
|
N/A |
|
|
List available and downloaded databases [False] |
|
N/A |
|
|
Downloads selected HMMs. Use the option |
|
|
|
|
Remove downloaded databases and FragGeneScan+ [False] |
|
N/A |
|
Input File Arguments:
Important
At least one sequence is required.
Accepted formats: [.fastq, .fq, .fasta, .fa, .fna, .ffn, .faa]
Example:
metacerberus.py --prodigal file1.fasta
metacerberus.py --config file.configIf a sequence is given in [.fastq, .fq] format, one of
--nanopore,--illumina, or--pacbiois required.:Option format interpretation:
--setup= accepts no additional options
--download DOWNLOAD= accepts one option, (represented by capitalized command ‘DOWNLOAD’)
--fraggenescan FRAGGENESCAN [FRAGGENESCAN...]= accepts one or greater options (represented by capitalized commands)
Output options:
Argument/Option |
Function [DEFAULT] |
Usage Format |
Accepted format |
# Options Accepted |
Example (Type as one line) |
|
path to output directory, defaults to “results-metacerberus” in current directory. [./results-`] |
|
output file path |
1 |
|
|
Flag to replace existing files. [False] |
|
|
N/A |
|
|
Flag to keep temporary files. [False] |
|
|
N/A |
|
|
Temp directory for RAY (experimental) [system tmp dir] |
|
|
1 |
|
Database options:
Argument/Option |
Function [DEFAULT] |
Usage Format |
Accepted format |
# Options Accepted |
Example (Type as one line) |
|
A list of databases for HMMER. Use the option |
|
|
=>1 |
|
|
Path to folder of databases [Default: under the library path of metacerberus] |
|
path to databases folder |
1 |
|
Optional Arguments:
Argument/Option |
Function [DEFAULT] |
Usage Format |
Accepted format |
# Options Accepted |
Example (Type as one line) |
|
Metagenomic nucleotide sequences (for prodigal) [False] |
|
|
N/A |
|
|
Sequences are treated as scaffolds [False] |
|
|
N/A |
|
|
Score cutoff for parsing HMMER results [60] |
|
whole integer value |
1 |
|
|
E-value cutoff for parsing HMMER results [1e-09] |
|
E-value |
1 |
|
|
Skip decontamination step. [False] |
|
|
N/A |
|
|
Skip PCA. [False] |
|
|
N/A |
|
|
Number of CPUs to use per task. System will try to detect available CPUs if not specified [Auto Detect] |
|
whole integer value |
1 |
|
|
Split files into smaller chunks, in Megabytes [Disabled by default] |
|
whole integer value |
1 |
|
|
Group multiple fasta files into a single file before processing. When used with –chunker (see above) can improve speed |
|
|
N/A |
|
|
show the version number and exit |
|
|
N/A |
|
|
show this help message and exit |
|
|
N/A |
|
|
FASTA File containing adapter sequences for trimming |
|
FASTA file |
1 |
|
|
FASTA File containing control sequences for decontamination |
|
FASTA file |
1 |
|
Note
Arguments/options that start with -- can also be set in a config file (specified via -c). Config file syntax allows: key=value, flag=true, stuff=[a,b,c] (for details, see syntax. In general, command-line values override config file values which override defaults.
Outputs (/final folder)
GAGE/Pathview
After processing the HMM files, MetaCerberus calculates a KO (KEGG Orthology) counts table from KEGG/FOAM for processing through GAGE and PathView.
GAGE is recommended for pathway enrichment followed by PathView for visualizing the metabolic pathways. A “class” file is required through the
--classoption to run this analysis.
For example (class.tsv):
Sample |
Class |
1A |
rhizobium |
1B |
non-rhizobium |
The output is saved under the step_10-visualizeData/combined/pathview folder. Also, at least 4 samples need to be used for this type of analysis.
GAGE and PathView also require internet access to be able to download information from a database.
MetaCerberus will save a bash script ‘run_pathview.sh’ in the step_10-visualizeData/combined/pathview directory along with the KO Counts tsv files and the class file for running manually in case MetaCerberus was run on a cluster without access to the internet.
Multiprocessing MultiComputing with RAY
MetaCerberus uses Ray for distributed processing. This is compatible with both multiprocessing on a single node (computer) or multiple nodes in a cluster.
MetaCerberus has been tested on a cluster using Slurm.
Example command to run your slurm script:
sbatch example_script.sh
Example Script:
#!/usr/bin/env bash
#SBATCH --job-name=test-job
#SBATCH --nodes=3
#SBATCH --tasks-per-node=1
#SBATCH --cpus-per-task=16
#SBATCH --mem=128MB
#SBATCH -e slurm-%j.err
#SBATCH -o slurm-%j.out
#SBATCH --mail-type=END,FAIL,REQUEUE
echo "====================================================="
echo "Start Time : $(date)"
echo "Submit Dir : $SLURM_SUBMIT_DIR"
echo "Job ID/Name : $SLURM_JOBID / $SLURM_JOB_NAME"
echo "Node List : $SLURM_JOB_NODELIST"
echo "Num Tasks : $SLURM_NTASKS total [$SLURM_NNODES nodes @ $SLURM_CPUS_ON_NODE CPUs/node]"
echo "======================================================"
echo ""
# Load any modules or resources here
conda activate metacerberus
# source the slurm script to initialize the Ray worker nodes
source ray-slurm-metacerberus.sh
# run MetaCerberus
metacerberus.py --prodigal [input_folder] --illumina --dir_out [out_folder]
echo ""
echo "======================================================"
echo "End Time : $(date)"
echo "======================================================"
echo ""
DESeq2 and Edge2 Type I errors
Both edgeR and DeSeq2 R have the highest sensitivity when compared to other algorithms that control type-I error when the FDR was at or below 0.1. EdgeR and DESeq2 all perform fairly well in simulation and via data splitting (so no parametric assumptions). Typical benchmarks will show limma having stronger FDR control across all types of datasets (it’s hard to beat the moderated t-test), and edgeR and DESeq2 having higher sensitivity for low counts (makes sense as limma has to filter these out / down-weight them to use the normal model on log counts). Further information about type I errors are present from Mike Love’s vignette here.
Contributing to MetaCerberus and Fungene
MetaCerberus as a community resource as recently acquired FunGene, we welcome contributions of other experts expanding annotation of all domains of life (viruses, bacteria, archaea, eukaryotes). Please send us an issue on our MetaCerberus GitHub open an issue; or email us we will fully annotate your genome, add suggested pathways/metabolisms of interest, make custom HMMs to be added to MetaCerberus and FunGene.
Copyright
This is copyrighted by University of North Carolina at Charlotte, Jose L Figueroa III, Eliza Dhungal, Madeline Bellanger, Cory R Brouwer and Richard Allen White III. All rights reserved. MetaCerberus is a bioinformatic tool that can be distributed freely for academic use only. Please contact us for commerical use. The software is provided “as is” and the copyright owners or contributors are not liable for any direct, indirect, incidental, special, or consequential damages including but not limited to, procurement of goods or services, loss of use, data or profits arising in any way out of the use of this software.
Citing MetaCerberus
If you are publishing results obtained using MetaCerberus, please cite:
Publication
Figueroa III JL, Dhungel E, Bellanger M, Brouwer CR, White III RA. 2024. MetaCerberus: distributed highly parallelized HMM-based processing for robust functional annotation across the tree of life. Bioinformatics.
Pre-print
Figueroa III JL, Dhungel E, Brouwer CR, White III RA. 2023. MetaCerberus: distributed highly parallelized HMM-based processing for robust functional annotation across the tree of life. bioRxiv.
Contact Us
The informatics point-of-contact for this project is Dr. Richard Allen White III. If you have any questions or feedback, please feel free to get in touch by email.
Or open an issue.